- Hartnup-Syndrom
-
Klassifikation nach ICD-10 E72.0 Störungen des Aminosäuretransportes ICD-10 online (WHO-Version 2006) Klassifikation
Hartnup-Krankheit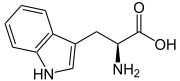
Tryptophan Die Hartnup-Krankheit (Hartnup-Syndrom) ist eine angeborene Störung des Transportes von Aminosäuren durch die Körperzellen.
Inhaltsverzeichnis
Epidemiologie
Die Krankheit wird autosomal-rezessiv vererbt. Mehrere Autoren wie z. B. Tahmoush, Kimmig. berichten aber auch über leichtere Erkrankungen wie Lichtdermatosen und Aminosäureurie bei nahen Verwandten mit wahrscheinlich nur einem defekten Gen. Die Erstmanifestation ist im Kindesalter zwischen drei und neun Jahren. Kann auch früher [1] oder erst im Erwachsenen auftreten.[2] [3]
Sie ist selten und tritt mit einer Prävalenz von lediglich 1:24.000 auf. Bei weitem nicht alle Träger des entsprechenden Gendefekts entwickeln diese Krankheit. Auffallend ist der extreme Unterschied im Verlauf bei durch Urin-Screenings gefundenen Personen und jenen, die erst durch einen akuten Ausbruch der Erkrankung gefunden wurden. Bei Ersteren wurden nur leichtere Erkrankungen beobachtet, viele erkrankten gar nicht. Bei der Zweiten ist die Sterblichkeitsrate hoch [4] [5] Es gibt mindestens zehn Mutationen von SLC6A19 in Chromosom 5p15.33, die den Transport unterschiedlich beeinträchtigen. Die meisten Betroffenen haben zwei unterschiedliche Mutationen. Die Allele D173N kommt bei 42% der durch Urin-Screening bei Neugeborenen in Amerika, Australien und Kanada gefundenen Genträger vor. [6] Gemeinsamkeiten der D173N Träger legen einen gemeinsamen Vorfahren vor mehr als Tausend Jahren nahe. Die Autoren postulieren, dass der lange Fortbestand dieser Allele auf ihrer Unfähigkeit den Transport vollständig zu deaktivieren beruht. Bei manchen Mutationen wie den Allelen D173N und P265L ist die Anzahl der Transportkanäle abhängig von ACE2 im Darm bzw Collectrin in den Nieren.[7]
Naturgemäß ist es bei der ersten Gruppe sehr viel einfacher Daten über eine relativ große Anzahl von Betroffenen zu erhalten. als Daten über die „wilden“ Fälle zu sammeln. Diese Daten beziehen sich jedoch auf Menschen bei denen lebenslänglich ein erhöhter Bedarf an Vitaminen und Proteinen berücksichtigt wurde, was neben der D173N Allele eine mögliche Erklärung für die milden Verläufe bietet. Dies führte auch weltweit in Fachbüchern und Veröffentlichungen zu einer Darstellung von Hartnup als „benign“.
Pathogenese
Es handelt sich um eine Störung des Transportes von Aminosäuren durch die Membranen der Körperzellen. Dafür verantwortlich ist der Mangel eines bestimmten kanalartigen Membranproteins (neutraler Aminosäuretransporter), welches für die Durchschleusung von aromatischen und neutralen Aminosäuren durch die Zellen zuständig ist. Der Defekt ist allgegenwärtig (ubiquitär), tritt aber in Geweben die verstärkt für die Resorption (Aufnahme) von Aminosäuren konzipiert sind, deutlicher in Erscheinung. Die Störung betrifft hauptsächlich den oberen Dünndarmabschnitt (Jejunum) und die Niere (proximaler Tubulus).
Durch das Unvermögen der Niere bestimmte Aminosäuren im Blut zu halten (verminderte Rückresorption), sammeln sich diese im Harn, und gehen den Körper folglich verloren. Im Urin finden sich Alanin, Serin, Threonin, Valin, Leucin, Isoleucin, Phenylalanin, Tyrosin, Tryptophan, Glutaminsäure, Asparagin und Histidin. Im selben Ausmaß, in dem der Verlust dieser Aminosäuren über den Harn geschieht, findet sich eine Resorptionsstörung derselben Substanzen im Dünndarm. Ein Mangel dieser Stoffe im Körper ist die logische Konsequenz. Der größte Teil der für den Lebenserhalt notwendigen essentiellen Aminosäuren wird durch Aminosäure-Recycling im Darm gewonnen. Jede Sekunde stirbt eine große Anzahl von Zellen, diese werden zerlegt und die Bestandteile in den Darm ausgeschieden. Bei der Hartnup-Krankheit ist dies jedoch eine Einbahnstraße wodurch der Bedarf einer äußeren Zufuhr der von dem Transportdefekt betroffenen Aminosäuren stark ansteigt bis zu einem Ausmaß, das durch bloße Nahrungsaufnahme nicht befriedigt werden kann. Auch normalerweise für Menschen nicht essentielle Aminosäuren können dadurch essentiell werden
Nun besitzt der menschliche Körper die Möglichkeit die fehlenden Substanzen weitgehend selbst herzustellen, und kann damit das Defizit auch weitgehend kompensieren. Als problematisch erweist sich jedoch der Mangel an Tryptophan. Dies ist eine essentielle Aminosäure. Das bedeutet, dass es für dieses Molekül keine Möglichkeit zur Synthese im menschlichen Stoffwechsel Intermediärmetabolismus) gibt. Diese Substanz muss unbedingt von Außen zugeführt werden.
Ein Mangel an Tryptophan kann begrenzt durch eine Substitution mit dessen Folgeprodukte Niacin und Nicotinamid beseitigt werden. Individuell unterschiedlich werden 1/80 bis 1/40 des Tryptophans in Nicotinamid umgewandelt. Durch eine erhöhte Zufuhr von Nicotinamid muss der Körper weniger Tryptophan umwandeln, wodurch mehr für andere Zwecke zur Verfügung steht.
Nicht resorbiertes Tryptophan wird im Darm durch Bakterien zu diversen Indolverbindungen metabolisiert. Diese werden im Gegensatz zum Tryptophan sehr wohl vom Darm resorbiert. Außerdem können sie die Blut-Hirn-Schranke überwinden. Viele zentralnervöse Symptome werden darauf zurückgeführt.
Symptomatik
Das Krankheitsbild gleicht im wesentlichen dem der Pellagra (Niacinmangel). Auslösende Faktoren für einen Krankheitsschub sind Fieber oder einige fiebersenkende Medikamente, starke Sonnenexposition, Stress und bestimmte Medikamente (Sulfonamide), Inhibitoren des Vitamin B Komplexes insbesondere von (Niacin), (Riboflavin) und (Pyridoxin). [8] Pellagra kann auch zu Störungen im Porphyrinstoffwechsel führen. [9] Porphyrieauslösende Medikamente können diese Störungen verstärken und schließlich einen Hartnup-Schub auslösen.
- Haut: Kennzeichnend für dieses Syndrom sind Hautläsionen in Form von erythematösen Ekzemen.
- Darm: Es kommt zu wiederkehrenden Durchfällen (Diarrhö).
- ZNS: Intermittierend finden sich neurologische oder psychiatrische Symptome. Es kann zu Störung der Bewegungskoordination (Ataxie), (Parkinsonoid), Lähmungen (Paresen) oder Stimmungschwankungen kommen. Selten werden auch demenzähnliche Zustände, Schielen (Strabismus) und Krampfanfälle (Konvulsionen) beschrieben.
Diagnose
Jeder Patient, der an pellagraartigen Symptomen erkrankt und nicht der für diese Krankheit typische ernährungsbedingte Niacinmangel nachvollziehbar ist, sollte auf das Vorhandensein des Gendefekts für die Hartnup-Krankheit untersucht werden.
Die Diagnose der Hartnup-Krankheit kann meist mittels Urinanalyse durchgeführt werden. Man findet häufig eine neutrale Aminoazidurie (Vorhandensein von Aminosäuren im Harn). Durch eine nicht vorhandene Aminoazidurie kann Hartnup jedoch nicht ausgeschlossen werden, wenn von einer ausreichenden Ernährung ausgegangen werden kann.[10] [11] [12]
Durch den Transportdefekt kann auch bei normalen Blutwerten ein Mangel in den Zellen bestehen. Paradoxerweise können durch die eingeschränkte Aufnahmefähigkeit der Zellen Blutwerte der betroffenen Aminosäuren sogar erhöht sein, wenn auch bei weitem nicht so stark wie z. B. bei Hypertryptophanämie.
Bei fehlender Aminoazidurie und scheinbar normalen Blutwerten kann ein einfacher Test auf Porphyrogene (Indolylacrylsäure und nahe Verwandte, Absorbtionsmaximum von 509 mµ) einen weiteren Hinweis geben. [13] In Morgenurin etwas Salzsäure geben (PH 2 - 2.5) und dann erwärmen (längere Zeit in die Sonne stellen tut es auch). vorhandene Porphyrogene bilden dann einen roten Farbstoff, durch den sich der Urin nahezu schwarz verfärben kann. Dieser Test kann auch bei Porphyrie und einigen anderen Erkrankungen, sowie nach Einnahme von Phenothiazinderivaten, positiv sein
Referenzbereich Tryptophan im Plasma oder Serum [14] Säuglinge: > 25 µmol/l< 69 µmol/l ~ > 0,5 mg/dl< 1,3 mg/dl. Kinder: > 32 µmol/l< 79 µmol/l ~ > 0,6 mg/dl < 1,6 mg/dl. Erwachsene: > 34µmol/l < 90 µmol/l ~ > 0,7 mg/dl < 1,7 mg/dl.Von einigen Laboren werden geradezu absurd niedrige untere Grenzwerte angegeben, bis hinunter zu 10 µmol/l. Sollte der Arzt zuerst den richtigen Verdacht gehabt haben, können solche Werte ein Menschenleben kosten.
Therapie
Die Behandlung besteht in der Verabreichung der fehlenden Substanzen (Substitutionstherapie). Eine tägliche Nahrungsergänzung von 50 bis 250 mg Nicotinamid verbessert die Situation bedeutend.
Außerdem empfiehlt sich eine proteinreiche Diät mit hohem Anteil an Tryptophan. Dieser findet sich hauptsächlich in Milch und Milchprodukten. Ferner findet sich Tryptophan in Geflügel, Rindfleisch, Nüssen und Kartoffeln.
In schweren Fällen ist es nicht möglich den Blutspiegel, der von dem Transportdefekt betroffenen Aminosäuren, mittels einer mit Proteinen angereicherten Diät signifikant zu erhöhen. Der Mangel muss dann durch Umgehung des Transportdefekts mittels intravenöser Substitution oder orale Substitution mit chemisch abgewandelten Aminosäuren beseitigt werden.[15] Geschieht dies nicht führt der Mangel zum Tod.
Die Pharmaindustrie bietet keine für eine Dauerbehandlung schwerer Fälle geeigneten Medikamente an. Mit einigem Aufwand sind aber auch schwere Fälle gut behandelbar. Auch wegen großen individuelen Unterschieden müssen Medikamente (Infusionen oder/und Ester) extra hergestellt werden. Eigentlich müsste kein Patient an Hartnup sterben.
Die Photodermatosen sind mit einer harnstoffhaltigen (10% - 15%) Salbe gut behandelbar, auch bei Blasenbildung.
Historisches
Diese Krankheit wurde erstmals 1956 in London bei Kindern der Familie Hartnup beobachtet. Angeblich litt auch die julisch-claudische Imperatorenfamilie von Julius Caesar bis hin zu Kaiser Nero an dieser Krankheit.
Literatur
- Dennis L. Kasper (Hrsg.), Eugene Braunwald (Hrsg.), Anthony Fauci: Harrison’s Principles of Internal Medicine. 16th Edition Volume II ISBN 0-07-139142-8
Einzelnachweise
- ↑ K. Schmidtke et al: Hartnup syndrome, progressive encephalopathy and allo-albuminaemia. A clinico-pathological case study. Eur J Pediatr. 1992 Dec;151(12):899-903.
- ↑ E. Mori et al.: Adult-onset Hartnup disease presenting with neuropsychiatric symptoms but without skin lesions. Rinsho Shinkeigaku. 1989 Jun;29(6):687-92.
- ↑ Oakley A, Wallace J.: Hartnup disease presenting in an adult. Clin Exp Dermatol. 1994 Sep;19(5):407-8.
- ↑ KH Daute et al.: The Hartnup syndrome. Report on a fatal course of the disease. Z Kinderheilkd. 1966 Jan 17;95(2):103-13.
- ↑ K. Schmidtke et al: Hartnup syndrome, progressive encephalopathy and allo-albuminaemia. A clinico-pathological case study. Eur J Pediatr. 1992 Dec;151(12):899-903.
- ↑ Persistence of the common Hartnup disease D173N allele in populations of European origin. Azmanov DN, Rodgers H, Auray-Blais C, Giguère R, Bailey C, Bröer S, Rasko JE, Cavanaugh JA. Ann Hum Genet. 2007 Nov;71(Pt 6):755-61. Epub 2007 Jun 7.
- ↑ Tissue-Specific Amino Acid Transporter Partners ACE2 and Collectrin Differentially Interact With Hartnup Mutations. Camargo SM, Singer D, Makrides V, Huggel K, Pos KM, Wagner CA, Kuba K, Danilczyk U, Skovby F, Kleta R, Penninger JM, Verrey F. Gastroenterology. 2008 Oct 29.
- ↑ Vitamin-Lexikon: Bässler, Grühn, Loew, Pietrzik
- ↑ Pschyrembel® Klinisches Wörterbuch Ausgabe 253
- ↑ Tahmoush AJ, Alpers DH, Feigin RD, Armbrustmacher V, Prensky AL: Hartnup disease. Clinical, pathological, and biochemical observations. In:Arch Neurol. 1976 Dec;33(12):797-807
- ↑ Freundlich E, Statter M, Yatziv S: Familial pellagra-like skin rash with neurological manifestations. In: Arch Dis Child. 1981 Feb;56(2):146-8
- ↑ Da Gloria ER, Assunção JG, Costa MA: Clinical picture of Hartnup disease. Without urine amino acids or any other identified metabolic disorder (a new entity). In: Med Cutan Ibero Lat Am. 1990;18(4):227-31 (Artikel auf Portugiesisch)
- ↑ J. Kimmig Hamburg: Stoffwechselstörungen bei polymorphen Lichtdermatosen und neuere Untersuchungsergebnisse im Zusammenhang mit dem Hartnup-Syndrom
- ↑ Lehrbuch der Klinischen Chemie und Pathobiochemie, Schattauer 3. Auflage
- ↑ Circumvention of defective neutral amino acid transport in Hartnup disease using tryptophan ethyl ester. A J Jonas and I J Butler J Clin Invest. 1989 July; 84(1): 200–204. PMCID: 303970
Bitte beachte den Hinweis zu Gesundheitsthemen!

Wikimedia Foundation.