- Neurofibromatose Typ 2
-
Klassifikation nach ICD-10 D33.3 Akustikusneurinom ICD-10 online (WHO-Version 2011)  NF-2 Locus
NF-2 Locus
Die Neurofibromatose Typ II (NF II), auch als zentrale Neurofibromatose bezeichnet, ist eine erbliche Tumorerkrankung. Ihr Hauptmerkmal ist das Vorkommen von gutartigen Hirntumoren, die sich symmetrisch im Bereich beider Hör- und Gleichgewichtsnerven entwickeln. Die meisten Patienten mit dieser Erkrankung leiden auch an Veränderungen der Augen. Ursache der NF II sind Mutationen eines Gens, das vermutlich Einfluss nimmt auf Form und Wanderungsverhalten bestimmter Zelltypen. Da die NF II genetisch bedingt ist, ist eine Heilung nicht möglich. Die Behandlung besteht in der Entfernung von Tumoren im Bereich des Gehirns und Rückenmarkes, sowie operativer Eingriffe im Bereich der Augen und der betroffenen Hirnnerven.
Die NF II ist etwa zehnmal seltener als die häufigste Form der Neurofibromatose, die periphere Neurofibromatose Typ 1 (Morbus Recklinghausen).
Inhaltsverzeichnis
Geschichte
Die Erstbeschreibung der Erkrankung stammt aus den 20er und 30er Jahren des 20. Jahrhunderts. 1933 beschrieben Gardner und Frazie eine Familie bei der sich über fünf Generationen in 38 Fällen eine Taubheit fand, die durch beidseitige Tumoren des Hörnervs verursacht waren. Zudem erblindeten 15 der beschriebenen Patienten. [1] Weitere Erkrankungsfälle wurden von Worster-Drought im Jahre 1937[2], von Feiling und Ward im Jahre 1920[3] und von Moyes im jahre 1968 [4] berichtet. In der Arbeit von Forster-Drought wiesen die Autoren auf eine Beschreibung von Wishart aus dem Jahre 1822 hin. Dieser beschrieb die Erkrankung erstmals.[5] Wishart war Präsident des Royal College of Surgeons of Edinburgh. Er beschrieb einen 21 Jahre alten Mann (Michael Blair) der unter einer beidseitigen Taubheit litt. Er zeigte eine besondere Kopfform und war seit dem vierten Lebensmonat rechtsseitig erblindet. Nach dem Tod des Patienten zeigten sich bei der Autopsie Tumoren des Gehirns und der Hirnhäute. Bemerkenswerterweise beschrieb Wishart einen „Tumor von der Größe einer kleinen Nuss, der sehr hart war und beidseitig am Hörnerv zu finden war, genau an der Stelle des Meatus acusticus internus“.
Inzidenz, Erbgang, Epidemiologie
Die NF II ist eine erbliche Tumorerkrankung mit autosomal dominantem Erbgang. Die betroffenen Menschen entwickeln bestimmte Tumoren des Nervensystems. Die Inzidenz (Erkrankungshäufigkeit) beträgt 1:35.000. Das klinische Erscheinungsbild ist vielfältig, aber alle Menschen mit dieser Erkrankung haben Mutationen am gleichen Genort. Betroffen ist ein Gen auf dem Chromosom 22. Bei etwa der Hälfte der Patienten liegt eine Neumutation vor.
Pathogenese, Molekularbiologie und pathophysiologische Zusammenhänge
Das bei NF2 veränderte Gen kodiert für ein Protein namens Merlin (Protein). Es wird einerseits vermutet dass Merlin, aufgrund seiner strukturellen Verwandtschaft Signale der Zellvermehrung an oder um die Plasmamembran hemmt. 2010 konnte gezeigt werden dass Merlin aber auch im Zellkern akkumuliert und E3-Ubiquitin-Ligase CRL4 hemmt und somit einen Signalweg anstößt, der sich gegen die Mitose der Zelle richtet. Bei NF2 liegt eine Mutation des Gens vor, welches den antimitotischen Effekt aufhebt.[6]
Pathologie
 Bilaterale Schwannome
Bilaterale SchwannomeDas sogenannte Akustikusneurinom bei der Neurofibromatose Typ II ist in Wirklichkeit ein Schwannom des Nervus vestibularis (Gleichgewichtsnerv). An dem falschen Begriff wird trotz besseren Wissens in der ganzen wissenschaftlichen Literatur festgehalten. Die vestibulären Schwannome wachsen langsam am inneren Eingang (Schädelbasisseite) des Meatus acusticus internus. Sie entstehen aus der Nervenscheide des oberen Abschnittes des N. vestibularis am Übergang des zentralen zum peripheren Myelin (sog. Redlich-Obersteiner-Zone) im Bereich des Porus acusticus internus, rund 1 cm entfernt vom Hirnstamm.
Krankheitsbild
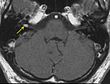 Schwannom des N. Vestibularis
Schwannom des N. VestibularisDas klinische Spektrum der Erkrankung ist breit. Mit dem Begriff klinisches Spektrum meint man das Auftreten von Symptomen, die im ursächlichen Zusammenhang mit der Erkrankung stehen.
- Hörnerv: Wie schon erwähnt findet man bei 90 % der Betroffenen bei einer Kernspintomographie des Schädels ein beidseitiges Akustikusneurinom.
 Intramedulläre Ependymome
Intramedulläre Ependymome- Rückenmark: Ebenso häufig finden sich spinale (das Rückenmark betreffende) Raumforderungen (in der Medizin bezeichnet der Begriff Raumforderung in vielen Fällen eine Gewebswucherung). Bei den Patienten mit einer NF Typ II werden die cranialen (den Schädel betreffenden) Raumforderungen fast immer symptomatisch (z.Bsp. Hörminderung). Die spinalen Raumforderungen werden aber nur in ca. 40 % der Fälle symptomatisch. Die spinalen Tumoren werden in zwei Gruppen unterteilt. Einmal findet man intramedulläre Raumforderungen. Damit sind Tumoren gemeint, die sich in der Gewebssubstanz des Rückenmarkes finden. Hier findet man vor allem Astrozytome und Ependymome. Zum anderen finden sich extramedulläre Raumforderungen. Hiermit meint man Tumoren oder Gewebsveränderungen, die sich im Rückenmarkskanal, aber außerhalb der Rückenmarkssubstanz befinden. Der extramedulläre Raum ist der Spalt zwischen der Oberfläche der Rückenmarkssäule und der knöchernen Wand des Rückenmarkskanales innerhalb der Wirbelsäule. In diesen Fällen treten vornehmlich Schwannome und Meningeome auf.
 Meningeome bei NF II
Meningeome bei NF II- Andere Läsionen des Zentralnervensystems: Die Meningeoangiomatose ist eine Läsion des zentralen Nervensystems mit plaque-ähnlichem Wachstum von Zellen um einzelne Gefäße, die bei NF II auch multifokal auftreten können. Gliale Hamartien sind umschriebene atypische Zellcluster des Hirnparenchyms. Intrakranielle Kalzifikationen werden gehäuft bei NF II beobachtet.
- Andere Hirnnerven und Hirnhäute: Etwa 50 % der Patienten haben Tumoren im Bereich der Hirnnerven oder Meningeome. Schwannosen sind kleine Zellproliferationen der Nervenscheiden ohne vollständige Tumorausbildung. Sie finden sich bei der NF II bevorzugt an den Austrittstellen der Spinalnerven.
- Haut: Bei Kindern kann das Auftreten von Neurofibromen ein Hinweis sein für das Vorliegen einer Neurofibromatose Typ II.
- Augen: Systematische Untersuchungen von Patienten mit einer NF Typ II ergaben, dass über 90 % der Erkrankten Augenveränderungen haben. Die weitaus häufigste Augenveränderung bei der NF II ist die sogenannte juvenile (im Jugendalter auftretende) subcapsuläre Katarakt (eine Form der Linsentrübung).
Die klinischen Symptome (Beschwerden, die die Patienten selbst bemerken und dem Arzt berichten) einer Läsion des Nervus vestibulocochlearis (Hör- und Gleichgewichtsnerv) durch eine Raumforderung im Kleinhirnbrückenwinkel sind folgende: Hörminderung (98 %), Tinnitus (Ohrgeräusch) (70 %), Gleichgewichtsstörung (67 %), cerebelläre Ataxie (Gangunsicherheit), Kopfschmerzen (32 %) und Taubheit (32 %) und Lähmung (10 %) im Bereich des Gesichtsnerven.
Die klinischen Zeichen (Veränderungen, die die Patienten selbst nicht bemerken, die aber ein Arzt bei einer körperlichen Untersuchung feststellen kann) einer Läsion des Nervus vestibulocochlearis (Hör- und Gleichgewichtsnerv) durch eine Raumforderung im Kleinhirnbrückenwinkel sind folgende: Störung des Cornealreflexes (33 %), Nystagmus (26 %), Hypästhesie im Bereich des N. facialis (26 %).
Bei technischen Untersuchungen ergeben sich folgende Befunde: Neben den typischen Befunden der Bildgebung (Erweiterung des Porus acusticus internus (Öffnung des Gehörganges) im Knochenfenster des Nativ-CCT, Kontrastmittelaufnahme der Tumoren) findet man eine Eiweißvermehrung im Liquor und pathologische Befunde bei den akustisch evozierten Potentialen. Ein Nystagmus lässt sich auch mit Hilfe einer Elektronystagmographie mit calorischer Stimulation charakterisieren.
Krankheitsverlauf
Charakteristisch für den Verlauf der Erkrankung ist die Tatsache, dass die Akustikusneurinome bei Patienten mit einer NF Typ II schon im jungen Erwachsenenalter auftreten, während bei den sporadischen Formen (damit meint man das Auftreten des Akustikusneurinoms unabhängig vom Vorliegen einer erblichen Tumorerkrankung) das Erkrankungsalter viel höher liegt.
Bei der NF II werden zwei Verlaufsformen beschrieben: Der sogenannte Wishart-Phänotyp ist gekennzeichnet durch das Auftreten von multiplen cerebralen und spinalen Raumforderungen vor dem 20. Lebensjahr mit rascher Tumorprogression. Der Feiling-Gardner-Phänotyp ist gekennzeichnet durch einen Krankheitsbeginn nach dem 20. Lebensjahr mit singulären zentralen Tumoren und langsamer Tumorprogression.
Genotyp-Phänotyp-Korrelation
Viele Patienten mit einer Neurofibromatose Typ II wurden in den letzten Jahren im Rahmen von wissenschaftlichen Studien behandelt. Dabei wurde die Ausprägung der Erkrankung, ihr Verlauf und der Typ der Mutation genau untersucht. Solche Studien beschäftigen sich mithin mit der Phänotyp-Genotyp-Korrelation. Der Sinn solcher Untersuchungen besteht darin, die Frage zu beantworten, ob bestimmte Mutationstypen mit bestimmten Krankheitserscheinungen eng verbunden sind. Man möchte am liebsten schon bei einem noch klinisch unauffälligen Patienten mit großer Sicherheit den Krankheitsverlauf vorhersagen können, um die Therapie zu optimieren. Bei diesen Studien hat man Folgendes festgestellt:
- Die meisten mutierten NF II-Gene führen zu verkürzten NF II Peptiden (Schwannominen).
- Es gibt keine Häufungsstelle der Mutationen (Hot-Spots).
- Patienten mit einer Frameshiftmutation- oder Nonsense-Mutation oder Mutationen oberhalb von Exon 7 haben einen schweren Verlauf.
- Patienten mit einer Missensemutation oder einem somatischen Moskaikmuster haben einen milden Verlauf.
- Bei Patienten mit einer Mutation in den Spleiß-Akzeptor-Sequenzen gibt es keinen bevorzugten Verlaufstyp.
- Kleinere Mutationen (Punktmutationen etc.) haben möglicherweise keine klinischen Auswirkungen.
- Es gibt Fälle, in denen Patienten mit der gleichen Mutation unterschiedliche Verlaufsformen zeigen.
Diese Befunde sprechen dafür, dass für die Ausprägung des Krankheitsbildes der NF Typ II noch andere Gene oder Umweltfaktoren verantwortlich sind.
- Mütterlicher Vererbung der Krankheit und Familien mit bekannter Anzipation kann mit einem ausgeprägterem Krankheitsbild einhergehen.
Diagnose
Das Kern- oder Kardinalsymptom der Erkrankung sind die beidseitigen gutartigen Tumoren des Hörnervs (sog. bilaterale Akustikusneurinome). Durch dieses Symptom ist die Krankheit definiert.
Als diagnostische Kriterien für eine Erkrankung bezeichnet man die Symptome, bei deren Vorliegen die klinische Diagnose gestellt werden darf. Für das vorliegen einer definitiven NF II sind dies:
- Der Nachweis von bilateralen Akustikusneurinomen mittels bildgebender Verfahren.
- Ein Verwandter ersten Grades mit einer NF II und der Nachweis von Neurofibromen, Meningeomen, Gliomen, Schwannomen.
- Ein Verwandter ersten Grades mit einer NF II und der Nachweis einer juvenilen posterioren subcapsulären Katarakt (Linsentrübung im jugendlichen Alter).
Folgende Kriterien machen das Vorliegen einer NF II wahrscheinlich:
- Einseitiges Akustikusneurinom vor dem 30. Lebensjahr und ein Meningom, Schwannom, Gliom oder Linsentrübung.
- Mehrere Meningeome und ein Gliom, Linsentrübung oder Schwannom vor dem 30 Lebensjahr.
Therapie
Für die Therapie ist die frühzeitige Erkennung der Erkrankung wichtig, da bei der NF Typ II schon Jugendliche erkranken. Häufig treten Symptome wie eine Hörminderung bis zu 10 Jahre vor der korrekten Diagnosestellung auf. Die Akustikusneurinome können frühzeitig operiert werden, um die Funktion des Gesichtsnervs zu erhalten. Allerdings führt dies nur bei der Hälfte der Patienten zu einem Erfolg. Patienten mit dem Wishart-Phänotyp erleiden zudem häufig Rezidive (Wiederauftreten des Tumors nach einer Operation). Bei einer Lähmung des Gesichtsnervs können rekonstruktive Eingriffe durchgeführt werden. Beim sogenannten Lidloading werden kleine Magnete in die Lider des betroffenen Auges implantiert, um eine chronische Bindehautentzündung durch einen unvollständigen Lidschluss bei einer Gesichtsnervenlähmung zu vermeiden. Die meisten Patienten mit einer NF II leiden im Rahmen der Erkrankung an einer Katarakt (Linsentrübung). Augenärztliche Operationen ersetzen die getrübte Linse durch eine künstliche Linse. Hierdurch wird die Verminderung des Sehvermögens verbessert. Bei Risikopatienten (Kinder von Betroffenen) empfiehlt man jährliche Kontrolluntersuchungen in spezialisierten Zentren. Zur Behandlung gehört auch das vorbeugende Erlernen der Gebärdensprache bei Patienten, die ein hohes Risiko für eine vollständige Ertaubung haben.
Operative Therapie des Akustikusneurinoms
Prinzipiell gilt, dass eine Operation bessere Ergebnisse ergibt, als die Bestrahlungstherapie. Es gibt sechs verschiedene Operationstechniken für die Entfernung eines ACN. Drei davon kommen nur in Ausnahmefällen zur Anwendung. Das Operationsverfahren wird ausgewählt nach der Größe des Tumors und dem Hörvermögen des Patienten.
- Der suboccipitale Zugang (SO) wird von Neurochirurgen bevorzugt und erlaubt den Erhalt des Hörvermögens. Der Nachteil dieses Verfahrens ist die höhere postoperative Komplikationsrate.
- Der translabyrinthine Zugang (TL) wird eher von HNO-Ärzten bevorzugt. Das Hörvermögen der betroffenen Seite wird dabei zerstört. Dies wird in Kauf genommen, wenn das Resthörvermögen des Patienten vor der Operation schon sehr schlecht ist. Der Vorteil dieses Verfahrens besteht in der Schonung des Gesichtsnerves. Nachteilig ist die erhöhte Rate an Liquorfisteln.
- Der subtemporale Zugang oder Weg durch die mittlere Schädelgrube (MF) wird nur bei kleinen Tumoren gewählt. Die Chance das Hörvermögen zu erhalten ist gut.
Große und mittelgroße Tumoren (2 bis 4 cm) können alternativ suboccipital oder translabyrinthär operiert werden. Hier entscheidet das Ausmaß des Hörvermögens und der Allgemeinzustand des Patienten (bei schlechtem Hörvermögen: TL; bei gutem Allgemeinzustand: SO). Kleine Tumoren (kleiner als 2 cm Durchmesser) bei Patienten mit schlechtem Hörvermögen werden mittels TL operiert. Kleine Tumoren bei Patienten mit gutem Hörvermögen und einem lateral (von der Schädelmittellinie weg) liegendem Tumor werden mittels MF operiert. Liegt der Tumor mehr medial (zur Schädelmittellinie hin), wählt man den SO Zugang.
Quellen
Einzelnachweise
- ↑ Gardner WJ, Frazier CH: Hereditary bilateral acoustic tumors. In: J Hered Vol. 22, pg. 7-8. 1933
- ↑ Worster-Drought C, Dickson WEC, McMenemey WH: Multiple meningeal and perineural tumors with analogous changes in the glia and ependyma (neurofibroblastomatosis). in: Brain Vol. 60, pg. 85-117. 1937
- ↑ Feiling A, Ward E: A familial form of acoustic tumour. In: Brit Med J Vol. 1, pg. 496-497. 1920
- ↑ Moyes PD Familial bilateral acoustic neuroma affecting 14 members from four generations. In: J Neurosurg Vol. 29, pg. 78-82. 1968 PMID 5674095
- ↑ Wishart JH Case of tumours in the skull, dura mater, and brain. In: Edinburgh Med Surg J Vol. 18 pg. 393-397, 1822
- ↑ Wei Li, Liru You, Jonathan Cooper, Gaia Schiavon, Angela Pepe-Caprio et al. : Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4 in the Nucleus, Cell, Volume 140, Issue 4, 477-490, 19 February 2010, abstract verfügbar als html ; zuletzt abgerufen am 1. März 2010
Bücher
- Jackler R. und Brackman D.E. (2 Ed.): Neurotology. Mosby. Elsevier 2004.ISBN 0-323-01830-0.
- Raymond D. Adams (Ed.): Principles of Neurology. McGraw-Hill. New York 1997. ISBN 0-07-067439-6.
- Bruce O. Berg (Ed.): Principles of Child Neurology. McGraw-Hill. New York 1996. ISBN 0-07-005193-3.
- Josef Dudel (Hrsg.): Neurowissenschaften. Springer. Berlin 1996. ISBN 3-540-61328-5.
- Mark S. Greenberg: Handbook of Neurosurgery. Lakeland 1997. ISBN 0-9626384-5-5.
- Wolfgang Hennig: Genetik. Springer. Berlin 2002. ISBN 3-540-42958-1.
- Thomas Herdegen (Hrsg.): Klinische Neurobiologie. Spektrum. Heidelberg 1997. ISBN 3-8274-0069-4.
- Andrew H. Kaye and Edward R. Laws Jr (Ed.): Brain Tumors. An Encyclopedic Approach. Churchill Livingston. Edinburgh 1995. ISBN 0-443-04840-1.
- John G. Nicholls (Hrsg.): Vom Neuron zum Gehirn. Gustav Fischer. Stuttgart 1995. ISBN 3-437-20517-X.
- Olaf Rieß und Ludger Schöls (Hrsg.): Neurogenetik. Molekulargenetische Diagnostik neurologischer Erkrankungen. Springer. Berlin 1998. ISBN 3-540-63874-1.
- Lewis P. Rowland (Ed.): Merrits Textbook of Neurology. Williams and Wilkins. Baltimore 1995. ISBN 0-683-07400-8.
- T. Strachan und A.P. Read (Ed.): Molekulare Humangenetik. Spektrum. Heidelberg 1996. ISBN 3-8274-0039-2.
Fachartikel
- Baser ME et al.: Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet. 2004 Aug;75(2):231-9. PMID 15190457
- Baser ME et al.: Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet. 2002 Oct;71(4):715-23. Epub 2002 Aug 22. PMID 12235555
- Baser ME et al.: Presymptomatik diagnosis in neurofibromatosis 2 using genetic markers, neuroimaging und ocular examination. (1996) Neurology 47:1269-1277. PMID 8909442
- Evans DGR et al.: A genetic study of neurofibromatosis 2. (1992a) J Med Gent 29:841-846 und 847-852. PMID 1479598 und PMID 1479599
- Evans DGR et al.: A clinical study of neurofibromatosis 2. (1992b) QJM 304:603-218. PMID 1484939
- Kaiser-Kupfer MI et al.: The association of posterior capsular lens opacities with bilateral acoustic neuromas in patients with neurofibromatosis type 2. Arch Ophthalmol. 1989 Apr;107(4):541-4. PMID 2705922
- Kluwe L et al.: Identification of NF2 germ-line mutations and comparison with neurofibromatosis 2 phenotypes. Hum Genet. 1996 Nov;98(5):534-8. Erratum in: Hum Genet 1997 Feb;99(2):292. PMID 8882871
- Mautner VF et al.: Neurofibromatosis 2 in the pediatric age group. Neurosurgery. 1993 Jul;33(1):92-6. PMID 8355853
- Mautner VF et al.: Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. AJR Am J Roentgenol. 1995 Oct;165(4):951-5. Erratum in: AJR Am J Roentgenol. 1996 May;166(5):1231. PMID 7676998
- Mautner VF et al.: The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996 May;38(5):880-5; discussion 885-6. PMID 8727812
- Merel P et al.: Screening for germ-line mutations in the NF2 gene. Genes Chromosomes Cancer. 1995 Feb;12(2):117-27. PMID 7535084
- Parry DM et al.: Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994 Oct 1;52(4):450-61. PMID 7747758
- Rouleau G et al.: Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987 Sep 17-23;329(6136):246-8. PMID 2888021
- Rouleau G et al.: Alteration in a new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. (1993) Nature 363:515-521. PMID 8379998
- Sainz J et al.: High frequency of nonsense mutations in the NF2 gene caused by C to T transitions in five CGA codons. Hum Mol Genet. 1995 Jan;4(1):137-9. PMID 7711726
- Trofatter JA et al.: A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993 Mar 12;72(5):791-800. Erratum in: Cell. 1993 Nov 19;75(4):826. PMID 8453669
- Warren C, et al.: Identification of recurrent regions of chromosome loss and gain in vestibular schwannomas using comparative genomic hybridisation. J Med Genet. 2003 Nov;40(11):802-6. PMID 14627667
Wiki-Links
 Commons: Neurofibromatose Typ 2 – Sammlung von Bildern, Videos und Audiodateien
Commons: Neurofibromatose Typ 2 – Sammlung von Bildern, Videos und AudiodateienAkustikusneurinom, Phakomatose, Neurofibromatose, Neurofibromatose Typ 1, Neurokutane Erkrankung, Liquor cerebrospinalis, Innenohr, Kleinhirn, Genmutation, Mutation, Gen, Punktmutation.

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in der Neurologie
- Krankheitsbild in der Neurochirurgie
- Erbkrankheit






Wikimedia Foundation.